4 Mechanismus der Inaktivierung von G-Proteinen. B. strukturelle und funktionelle Organisation von G-Proteinen. Andere Immunglobulin-bindende Proteine
G-Proteine (GTP-bindende Proteine) sind universelle Vermittler bei der Signalübertragung von Rezeptoren zu Zellmembranenzymen, die die Bildung von Second Messenger des Hormonsignals katalysieren. G-Proteine sind Oligomere, die aus α-, β- und γ-Untereinheiten bestehen. Die Zusammensetzung der βγ-Dimere variiert geringfügig in verschiedenen Geweben, aber innerhalb derselben Zelle weisen alle G-Proteine in der Regel den gleichen Satz an βγ-Untereinheiten auf. Daher werden G-Proteine normalerweise anhand ihrer α-Untereinheiten unterschieden. . 16 Gene, die verschiedene α-Untereinheiten von G-Proteinen kodieren, wurden identifiziert. Einige der Gene enthalten aufgrund des alternativen RNA-Spleißens mehr als ein Protein.
Jede α-Untereinheit im G-Protein hat spezifische Zentren:
GTP- oder GDP-Bindung;
Wechselwirkungen mit dem Rezeptor;
Bindungen an βγ-Untereinheiten;
Phosphorylierung durch Proteinkinase C;
Wechselwirkungen mit dem Enzym Adenylatcyclase oder Phospholipase C.
Der Struktur von G-Proteinen fehlen α-helikale, membrandurchspannende Domänen. G-Proteine gehören zur Gruppe der „verankerten“ Proteine (Abb. 5-34).
Reis. 5-34. Position G-Proteine in der Membran. Für die Assoziation von G-Proteinen ist die Acylierung von α-Protomeren mit aliphatischen Resten von Fettsäuren, Myristinsäure (M) oder Isoprensäure, wichtig. Die γ-Untereinheit des G-Proteins verfügt über eine Geranyl-Geranyl-Gruppe (G), die über eine Thioetherbindung an einen C-terminalen Cysteinrest gebunden ist.
Aktivitätsregulierung G -Proteine
Es gibt eine inaktive Form des G-Proteins – den αβγ-GDP-Komplex und die aktivierte Form αβγ-GTP. Die Aktivierung des G-Proteins erfolgt durch Wechselwirkung mit dem Aktivator-Rezeptor-Komplex; eine Änderung der Konformation des G-Proteins verringert die Affinität der α-Untereinheit für das GDP-Molekül und erhöht sie für GTP. Der Ersatz von GDP durch GTP im aktiven Zentrum des G-Proteins stört die Komplementarität zwischen α-GTP- und βγ-Untereinheiten. Der mit dem Signalmolekül verbundene Rezeptor kann eine große Anzahl von G-Proteinmolekülen aktivieren und so in diesem Stadium für eine Verstärkung des extrazellulären Signals sorgen (Abb. 5-35).

Reis. 5-35. Zyklus der G-Protein-Funktion. R s – Rezeptor; G – Hormon; AC – Adenylatcyclase.
Die aktivierte G-Protein-α-Untereinheit (α-GTP) interagiert mit einem bestimmten Zellmembranprotein und verändert dessen Aktivität. Solche Proteine können die Enzyme Adenylatcyclase, Phospholipase C, cGMP-Phosphodiesterase, Na+-Kanäle, K+-Kanäle sein.
Die nächste Stufe des G-Protein-Funktionszyklus ist die Dephosphorylierung von GTP, das an die α-Untereinheit gebunden ist, und das Enzym, das diese Reaktion katalysiert, ist die α-Untereinheit selbst.
Die Dephosphorylierung führt zur Bildung eines α-GDP-Komplexes, der nicht zu einem spezifischen Membranprotein (z. B. Adenylatcyclase) komplementär ist, aber eine hohe Affinität zu den βγ-Protomeren aufweist. Das G-Protein kehrt in seine inaktive Form, αβγ-GDP, zurück. Mit der anschließenden Aktivierung des Rezeptors und dem Ersatz des GDP-Moleküls durch GTP wiederholt sich der Zyklus erneut. Somit transportieren die α-Untereinheiten von G-Proteinen ein stimulierendes oder hemmendes Signal von einem Rezeptor, der durch einen primären Botenstoff (z. B. ein Hormon) aktiviert wird, zu einem Enzym, das die Bildung eines sekundären Botenstoffs katalysiert.
Einige Formen von Proteinkinasen können die α-Untereinheiten von G-Proteinen phosphorylieren. Die phosphorylierte α-Untereinheit ist nicht komplementär zu einem bestimmten Membranprotein, beispielsweise Adenylatcyclase oder Phospholipase C, und kann daher nicht an der Signaltransduktion teilnehmen.
G. Adenylatcyclase
Das Enzym Adenylatcyclase, das die Umwandlung von ATP in cAMP katalysiert (Abb. 5-36), ist ein Schlüsselenzym im Signaltransduktionssystem der Adenylatcyclase. Adenylatcyclase kommt in allen Zelltypen vor.

Reis. 5-36. Bildung von zyklischem Adenosinmonophosphat (cAMP).
Das Enzym gehört zur Gruppe der integralen Proteine der Zellmembran; es verfügt über 12 Transmembrandomänen. Extrazelluläre Fragmente der Adenylatcyclase werden glykosyliert. Die zytoplasmatischen Domänen der Adenylatcyclase verfügen über zwei katalytische Zentren, die für die Bildung von cAMP verantwortlich sind, einem sekundären Botenstoff, der an der Regulierung der Aktivität des Enzyms Proteinkinase A beteiligt ist.
Die Aktivität der Adenylatcyclase wird sowohl durch extrazelluläre als auch intrazelluläre Regulatoren beeinflusst. Extrazelluläre Regulatoren (Hormone, Eicosanoide, biogene Amine) führen die Regulierung über spezifische Rezeptoren durch, die mit Hilfe von α-Untereinheiten von G-Proteinen Signale an die Adenylatcyclase übermitteln. αs – Untereinheit (stimulierend), wenn sie mit Adenylatcyclase interagiert, aktiviert sie das Enzym, αi-Untereinheit (hemmend) hemmt das Enzym. Adenylatcyclase wiederum stimuliert die Manifestation der GTP-Phosphatase-Aktivität von α-Untereinheiten. Durch die Dephosphorylierung von GTP werden die Untereinheiten a s -GDP und a i -GDP gebildet, die nicht komplementär zur Adenylatcyclase sind.
Von den 8 untersuchten Isoformen der Adenylatcyclase sind 4 Ca 2+ -abhängig (aktiviert durch Ca 2+). Die Regulierung der Adenylatcyclase durch intrazelluläres Kalzium ermöglicht es der Zelle, die Aktivitäten der beiden wichtigsten sekundären Botenstoffe cAMP und Ca 2+ zu integrieren.
D. Phospholipasen
Phospholipasen sind Enzyme der Hydrolase-Klasse, die den Abbau von Glycerophospholipiden katabolisieren. Es gibt sekretorische Phospholipasen, die Teil des Pankreassaftes sind, und zelluläre Phospholipasen. Die zellulären Phospholipasen A 1, A 2, D, C unterscheiden sich in ihrer Spezifität für die gespaltene Gruppe. Alle Phospholipasen sind kalziumabhängige Enzyme (Abb. 5-37).

Reis. 5-37. Wirkung von Phospholipasen.
Phospholipase C- ein Enzym, das die Phosphoesterbindung in Glycerophospholipiden hydrolysiert. In menschlichen Zellen wurden 10 Isoformen der Phospholipase C identifiziert, die sich in Molekulargewicht, Lokalisierung, Regulationsweise und Substratspezifität unterscheiden. In der Struktur aller Phospholipase-C-Isoformen fehlen hydrophobe Domänen, die ihre Wechselwirkung mit der Membran gewährleisten könnten. Einige Formen der Phospholipase C werden jedoch über einen hydrophoben „Anker“ – den Acylrest der Myristinsäure – oder durch Wechselwirkung mit der Oberfläche der Doppelschicht gebunden. Die katalytische Aktivität aller Phospholipase-C-Isoformen hängt von Calciumionen ab.
Die meisten Phospholipasen C sind spezifisch für Phosphatidylinositole und hydrolysieren praktisch keine anderen Arten von Phospholipiden. Das aktive Enzym kann bis zu 50 % der gesamten Phosphatidylinositole in der Zellmembran hydrolysieren. Bei der Hydrolyse von Phosphatidylinositol-4,5-bisphosphat (PIF 2) entstehen die Produkte Diacylglycerol (DAG) und Inositol-1,4,5-triphosphat (IP 3), die als sekundäre Botenstoffe bei der transmembranen Signaltransduktion entlang des Inositolphosphatwegs dienen.
E. Proteinkinasen
Alle polaren Signalmoleküle, die über Membranrezeptoren auf die Zielzelle einwirken, erfüllen ihre biologische Funktion durch die Phosphorylierung spezifischer Proteine und Enzyme, die den Stoffwechsel in der Zelle regulieren. Phosphorylierung verändert (erhöht oder verringert) ihre Aktivität. Proteinkinasen katalysieren die Phosphorylierung von Proteinen an den Aminosäureresten Serin, Threonin und Tyrosin. Proteinkinasen können eine Untereinheit eines Membranrezeptors sein, beispielsweise der Insulinrezeptor-Protein-Tyrosinkinase, deren Aktivität durch ein Hormon reguliert wird. Eine weitere Gruppe sind Proteinkinasen, die durch sekundäre hormonelle Signalbotenstoffe (cAMP, cGMP, Ca 2+, DAG) reguliert werden, beispielsweise Proteinkinase A, Proteinkinase C, Proteinkinase G, Calmodulin-abhängige Proteinkinasen usw.
Proteinkinase A
Proteinkinasen A (cAMP-stimuliert) sind am Signaltransduktionssystem der Adenylatcyclase beteiligt. Proteinkinase A besteht aus 4 Untereinheiten R 2 C 2 – zwei regulatorischen Untereinheiten (R 2) und zwei katalytischen (C 2) (siehe Abb. 5-41). Der R 2 C 2-Komplex besitzt keine enzymatische Aktivität.
Der R 2 C 2-Komplex wird auf unterschiedliche Weise an die Membran gebunden. Einige Formen der Proteinkinase A werden durch den aliphatischen Myristinsäurerest der katalytischen Untereinheiten „verankert“. In vielen Geweben ist Proteinkinase A mit dem „verankerten“ Protein AKAP s (aus dem Englischen) verbunden. cAMP-abhängige Proteinkinase-Ankerproteine). ACAP s verfügt über ein Bindungszentrum für die regulatorischen Untereinheiten der Proteinkinase A. Mit Hilfe des ACAP s-Proteins bindet Proteinkinase A an die Membran in dem Bereich, in dem sich Enzyme befinden, die die Bildung von cAMP (Adenylatcyclase) oder dessen Hydrolyse katalysieren ( Phosphodiesterase) sowie Proteine, an deren Aktivitätsregulation das Enzym beteiligt ist, beispielsweise spannungsgesteuerte Ca 2+-Kanäle.
Die regulatorischen Untereinheiten der Proteinkinase A verfügen über spezifische Bindungsstellen für cAMP. Die Anlagerung von cAMP an regulatorische Untereinheiten führt zu einer Konformationsänderung dieser und einer Abnahme der Affinität zu katalytischen Untereinheiten C, die Dissoziation erfolgt nach folgendem Schema:
cAMP 4 + R 2 C 2 -> cAMP 4 R 2 + C + C
Untereinheiten C sind die aktive Form der Proteinkinase A, die Phosphorylierungsreaktionen an Serin und Threonin katalysiert. Die katalytischen Untereinheiten C verschiedener Arten von Proteinkinasen sind nicht identisch; sie unterscheiden sich hauptsächlich in ihrer Spezifität gegenüber Substratproteinen.
Proteinkinase C
Proteinkinasen C sind am Signaltransduktionssystem von Inositolphosphat beteiligt. Das Enzym besteht aus zwei funktionell unterschiedlichen Domänen – der regulatorischen und der katalytischen. Der Regulierungsbereich enthält 2 Strukturen („Zinkfinger“) besteht aus Fragmenten der Peptidkette, ist reich an Cystein und enthält 2 Zinkionen (siehe Abschnitt 1). Zinkfinger sind an der Bindung von Diacylglycerin beteiligt. Ein weiteres Fragment der regulatorischen Domäne weist eine hohe Affinität zu Ca 2+ auf. Durch die Erhöhung der Calciumkonzentration im Zytosol erhöht sich die Affinität der Proteinkinase C zum Membranphosphatidylserin. Durch die Translokation der Proteinkinase C zur Membran kann das Enzym an DAG binden, was die Affinität der Proteinkinase C für Calciumionen weiter erhöht (Abb. 5-38). Die häufigsten Isoformen der Proteinkinase C werden durch Ca 2+ , Diacylglycerin und Phosphatidylserin aktiviert.

5-38. Regulierung der Proteinkinase C (PKC)-Aktivität. PS – Phosphatidylserin; DAG – Diacyglycerin.
Die katalytische Domäne verfügt über ein Zentrum, das ATP und das Substratprotein bindet. Das aktive Enzym Proteinkinase C phosphoryliert an Serin- und Threoninresten. Eine Abnahme der Calciumionenkonzentration in der Zelle stört die Verbindung der Proteinkinase C mit Phosphatidylserin und Diacylglycerin, das Enzym wird inaktiv und trennt sich von der Membran.
3. Proteinkinasen G
Im Gegensatz zu Proteinkinase A ist Proteinkinase G nicht in allen Geweben vorhanden; sie kommt in der Lunge, im Kleinhirn, in der glatten Muskulatur und in Blutplättchen vor. Proteinkinase-G-Isoformen können membrangebunden sein oder sich im Zytoplasma befinden. Die lösliche Proteinkinase C besteht aus zwei identischen Untereinheiten, die jeweils über zwei Bindungsstellen für cGMP verfügen. Die Bindung von cGMP an regulatorische Zentren führt zu Konformationsänderungen in den Untereinheiten und erhöht die katalytische Aktivität des Enzyms (Abb. 5-39). Proteinkinase G ist wie Proteinkinasen A und C spezifisch für bestimmte Proteinsubstrate, die sie an Serin- und Threoninresten phosphoryliert.

Reis. 5-39. Regulierung der Aktivität der Proteinkinase G (PKG).
UND Phosphodiesterase
Phosphodiesterasen sind Enzyme, die die Umwandlung von cAMP (Abb. 5-40) oder cGMP in die inaktiven Metaboliten AMP oder GMP katalysieren. Phosphodiesterasen reduzieren die Konzentration sekundärer Botenstoffe und unterbrechen die durch den Rezeptoraktivator verursachte Transformationskette.

Abb.5-40. Umwandlung von cAMP in AMP.
Phosphodiesterasen kommen in Gewebezellen in zwei Formen vor: lösliches Protein und membrangebunden. Membrangebundene Formen des Enzyms machen in verschiedenen Geweben 5–40 % aus. Im selben Gewebe können verschiedene Formen der Phosphodiesterase vorhanden sein, die sich in der Affinität zu Substraten, im Molekulargewicht, in der Ladung, in den regulatorischen Eigenschaften und in der Lokalisierung in der Zelle unterscheiden.
Zyklische Nukleotid-Phosphodiesterasen haben keine absolute Spezifität, daher ist in der Regel dieselbe Form des Enzyms in der Lage, sowohl cAMP als auch cGMP zu hydrolysieren. Allerdings können sich die Hydrolyseraten dieser beiden Nukleotide durch dieselbe Phosphodiesterase erheblich unterscheiden. Dies hängt davon ab, welche Phosphodiesterase in der Zelle vorhanden ist – spezifischer für cAMP oder spezifischer für cGMP, vom Verhältnis der Konzentrationen von cAMP und cGMP in der Zelle und von der Wirkung der Phosphodiesterase-Regulatoren.
Die meisten Gewebe enthalten Phosphodiesterase-1, die spezifischer für cAMP ist und durch Ca 2+, den 4 Ca 2+ -Calmodulin-Komplex und cGMP aktiviert wird.
Beim Senden eines Signals an Rezeptoren gekoppelt, praktisch der gleiche Grundmechanismus ist beteiligt. Die Bindung des Agonisten durch den Rezeptor führt zu einer Konformationsänderung des Rezeptorproteins. Diese Veränderung wird auf das G-Protein übertragen: Die α-Untereinheit wandelt Guanosindiphosphat (GDP) in Guanosintriphosphat (GTP) um, dissoziiert dann von den beiden anderen Untereinheiten, bindet an das Effektorprotein und ändert seinen Funktionszustand.
Prinzipiell sind die β- und γ-Untereinheiten auch in der Lage, mit Effektorproteinen zu interagieren, während die α-Untereinheit langsam hydrolysiert gebundenes BTP an das BIP. G a -GDP hat keine Affinität zum Effektorprotein und bindet erneut an die β- und γ-Untereinheiten. G-Proteine können entlang der Membran diffundieren; Sie sind nicht an einzelne Rezeptorproteine gebunden. Es besteht jedoch ein Zusammenhang zwischen Rezeptortypen und G-Proteintypen.
Außerdem, α-Untereinheiten einzelner G-Proteine unterscheiden sich in ihrer Affinität zu verschiedenen Effektorproteinen sowie in der Art ihrer Wirkung auf diese. G a -GTP des G s-Proteins stimuliert die Adenylatcyclase, während G a -GTP des G i-Proteins ihr Inhibitor ist.
Gruppe G-Protein gekoppelt Zu den Rezeptoren gehören muskarinische cholinerge Rezeptoren, Adrenorezeptoren von Noradrenalin und Adrenalin sowie Rezeptoren für Dopamin, Histamin, Serotonin, Glutamat, GABA, Morphin, Prostaglandine, Leukotriene und viele andere Mediatoren und Hormone.
Zu den wichtigsten Effektorproteinen an G-Protein-gekoppelten Rezeptoren Dazu gehören Adenylatcyclase (ATP => intrazellulärer Botenstoff cAMP), Phospholipase C (Phosphatidylinositol => intrazellulärer Botenstoff Inositoltriphosphat und Diacylglycerin) und Ionenkanalproteine. Eine Vielzahl zellulärer Funktionen wird durch die Konzentration von zellulärem zyklischem Adenosinmonophosphat (cAMP) reguliert, da cAMP die Aktivität der Proteinkinase A erhöht, die die Umwandlung von Phosphatgruppen in funktionelle Proteine katalysiert.
Förderung cAMP-Level führt zu einer Abnahme des Tonus der glatten Muskulatur, einer Erhöhung der Kontraktilität des Herzmuskels sowie einer Zunahme der Glykogenolyse und Lipolyse. Die Phosphorylierung kardialer Ca 2+ -Kanalproteine erhöht die Wahrscheinlichkeit einer Kanalöffnung während der Membrandepolarisation. Es ist zu beachten, dass cAMP durch Phosphodiesterase inaktiviert wird. Inhibitoren dieses Enzyms erhöhen die intrazelluläre Konzentration von cAMP und wirken wie Adrenalin.
Eiweiß Rezeptor Es wird unabhängig phosphoryliert und verliert die Fähigkeit, das gebundene Protein G zu aktivieren. Dies ist einer der Mechanismen, die dazu beitragen, die Empfindlichkeit der Zelle bei längerer Stimulation des Rezeptors mit einem Agonisten (Desensibilisierung) zu verringern.
Aktivierung Phospholipase C führt zur Hydrolyse des Membranphospholipids Phosphatidylinositol-4,5-diphosphat zu Inositoltriphosphat (IP 3) und Diacylglycerin (DAG). Inositoltriphosphat stimuliert die Freisetzung von Ca 2+ aus Speicherorganellen, was zur Kontraktion glatter Muskelzellen, zum Abbau von Glykogen oder zur Aktivierung der Exozytose führt. Diacylglycerin stimuliert die Proteinkinase C, die eine Reihe von Serin- oder Threonin-haltigen Enzymen phosphoryliert.
Manche G-Proteine bewirken die Öffnung von Kanalproteinen. Dadurch werden K+-Kanäle aktiviert (die Wirkung von ACh auf den Sinusknoten, die Wirkung von Opioiden auf die Übertragung von Nervenimpulsen).

Unter organischen Substanzen Eichhörnchen, oder Proteine sind die zahlreichsten, vielfältigsten und wichtigsten Biopolymere. Ihr Anteil ist 50 - 80% trockene Zellmasse.
Proteinmoleküle sind groß und werden deshalb so genannt Makromoleküle. Neben Kohlenstoff, Sauerstoff, Wasserstoff und Stickstoff können Proteine auch Schwefel, Phosphor und Eisen enthalten. Proteine unterscheiden sich voneinander in der Anzahl (von einhundert bis mehreren tausend), der Zusammensetzung und der Reihenfolge der Monomere. Proteinmonomere sind Aminosäuren (Abb. 1)
Durch unterschiedliche Kombinationen von allem entsteht eine unendliche Vielfalt an Proteinen 20 Aminosäuren. Jede Aminosäure hat ihren eigenen Namen, ihre besondere Struktur und ihre eigenen Eigenschaften. Ihre allgemeine Formel lässt sich wie folgt darstellen:
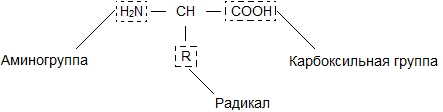
Ein Aminosäuremolekül besteht aus zwei Teilen, die mit allen Aminosäuren identisch sind, von denen einer die Aminogruppe ist ( -NH 2) mit basischen Eigenschaften, das andere mit einer Carboxylgruppe ( -COOH) mit sauren Eigenschaften. Teil eines Moleküls, genannt Radikal ( R), verschiedene Aminosäuren haben unterschiedliche Strukturen. Das Vorhandensein basischer und saurer Gruppen in einem Aminosäuremolekül bestimmt ihre hohe Reaktivität. Über diese Gruppen werden Aminosäuren zu Proteinen zusammengefasst. In diesem Fall entsteht ein Wassermolekül und die freigesetzten Elektronen bilden eine Peptidbindung. Deshalb werden Proteine genannt Polypeptide.
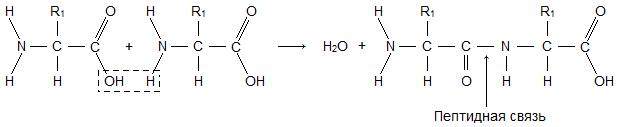
Proteinmoleküle können unterschiedliche räumliche Konfigurationen haben und in ihrer Struktur gibt es vier Ebenen der strukturellen Organisation.
Die Reihenfolge der Aminosäuren in einer Polypeptidkette ist Primärstruktur Eichhörnchen. Es ist für jedes Protein einzigartig und bestimmt dessen Form, Eigenschaften und Funktionen.
Die meisten Proteine haben aufgrund der Bildung von Wasserstoffbrückenbindungen zwischen ihnen eine helikale Form -CO- Und -NH- Gruppen verschiedener Aminosäurereste der Polypeptidkette. Wasserstoffbrückenbindungen sind schwach, aber zusammen ergeben sie eine ziemlich starke Struktur. Diese Spirale ist Sekundärstruktur Eichhörnchen.
Tertiärstruktur- dreidimensionale räumliche „Packung“ der Polypeptidkette. Das Ergebnis ist eine bizarre, aber spezifische Konfiguration für jedes Protein – Kügelchen. Die Festigkeit der Tertiärstruktur wird durch die verschiedenen Bindungen gewährleistet, die zwischen Aminosäureresten entstehen.
Quartärstruktur nicht typisch für alle Proteine. Es entsteht durch die Kombination mehrerer Makromoleküle mit Tertiärstruktur zu einem komplexen Komplex. Beispielsweise ist Hämoglobin im menschlichen Blut ein Komplex aus vier Proteinmakromolekülen.
Diese Komplexität der Struktur von Proteinmolekülen hängt mit der Funktionsvielfalt dieser Biopolymere zusammen.
Als Verletzung der natürlichen Struktur eines Proteins wird bezeichnet Denaturierung. Es kann unter dem Einfluss von Temperatur, Chemikalien, Strahlungsenergie und anderen Faktoren auftreten. Bei einer schwachen Wirkung zerfällt nur die Quartärstruktur, bei einer stärkeren die Tertiär- und dann die Sekundärstruktur, und das Protein verbleibt in Form einer Polypeptidkette.
Dieser Vorgang ist teilweise reversibel: Wenn die Primärstruktur nicht beschädigt wird, kann das denaturierte Protein seine Struktur wiederherstellen. Daraus folgt, dass alle Strukturmerkmale eines Proteinmakromoleküls durch seine Primärstruktur bestimmt werden.
Außer einfache Proteine, die nur aus Aminosäuren bestehen, gibt es auch komplexe Proteine
G – Proteine verstärken das übertragene Signal. Beispielsweise kann der Nervenimpulsgeber Noradrenalin in wenigen Millisekunden mit seinem Membranrezeptor interagieren. G-Protein erhöht die Dauer des Signals von Millisekunden auf mehrere zehn Sekunden, was äußerst wichtig ist (es besteht keine Notwendigkeit, ständig Signale an das Nervensystem zu senden). Es kommt zu einer Einsparung nervöser Energie.
G-Protein-gekoppelte Rezeptoren bilden die Familie der „Serpentinen“- (oder Schlangen-)Rezeptoren, die so genannt werden, weil ihre Polypeptidketten die Plasmamembran sieben Mal durchqueren.
Zu dieser Familie gehören Rezeptoren für adrenerge Amine, Serotonin, Acetylcholin (Muskarinsäure), viele Peptidhormone, Riechepithel und visuelle Rezeptoren (in Zapfen und Stäbchen der Netzhaut). Das Informationsmolekül (zum Beispiel Noradrenalin) bindet an die „Tasche“, die durch die Transmembranregionen des Rezeptors gebildet wird. Die daraus resultierenden Konformationsänderungen dieser Regionen werden auf die zytoplasmatischen Schleifen des Rezeptors übertragen, die das G-Protein aktivieren. Je mehr Agonistenmoleküle vorhanden sind, desto schneller bindet es an den Rezeptor.
Desensibilisierung von Rezeptoren.
Dies bedeutet, dass nach Erreichen eines anfänglich hohen Wirkungsniveaus (z. B. intrazelluläre cAMP-Akkumulation, Na + -Strom, Muskelkontraktion usw.) die Reaktion der Zelle über einen Zeitraum von Sekunden oder Minuten allmählich abnimmt, selbst trotz der ständigen Präsenz der Signalisierung Molekül. Die Desensibilisierung ist reversibel. Somit führt seine wiederholte Exposition 15 Minuten nach der Entfernung des Signalmoleküls zu einer Reaktion, deren Ausmaß mit der ursprünglichen Reaktion vergleichbar ist.
Down – Regulierung der Rezeptoren.
Wenn der Rezeptor überstimuliert wird, kann er in das Zytosol sinken und die Zelle „verdaut“ ihn mit Hilfe lysosomaler Enzyme in Aminosäuren. Die Membran, in der sich der Rezeptor befand, wird wiederhergestellt.
Ar – Regulierung.
Wenn die Nerven, die einen Muskel versorgen, operativ durchtrennt werden, empfängt der Muskel keine Signale vom Nervensystem und kann sich nicht zusammenziehen. Die Muskelreaktion auf die Denervierung zielt auf die Synthese zusätzlicher Rezeptoren ab. Sie werden synthetisiert und in die äußere Zellmembran eingebettet. Die Zelle möchte ein Signal zur Kontraktion erhalten. Das Signal kommt nicht an (der Nerv ist durchtrennt), obwohl es viele Rezeptoren gibt und diese besonders anfällig für den Neurotransmitter sind. Die Rezeptoren befinden sich sogar an anderen Stellen, weit entfernt von der Verbindung des Nervs mit dem Muskel. Dabei handelt es sich um die sogenannte AP – Rezeptorregulation – die Synthese neuer Rezeptoren durch die Zelle und deren Integration in die Membran. Rezeptoren werden ständig aktualisiert. Die Lebensdauer des Rezeptors beträgt mehrere Tage. Anstelle des von der Zelle Altgewordenen und Zerstörten baut sie ein Neues auf. Dies ist ein dynamischer Prozess.
Dadurch empfängt der in der Membran eingebaute Rezeptor ein Signal (Nervenimpuls, Arzneimittelhormon) und das G-Protein verstärkt dieses Signal. Das Effektorelement (Enzym) setzt dieses Signal um und löst so die Synthese von Second Messenger in der Zelle aus. Sie verändern die Geschwindigkeit biochemischer Reaktionen in Zellen und setzen die vom Nerven- oder Hormonsystem gesendeten Signale direkt um.
Sekundäre Vermittler.
1) Lager. ist an der Übertragung hormoneller Wirkungen beteiligt wie: 1) Mobilisierung von Energiereserven (Abbau von Kohlenhydraten in der Leber oder Triglyceriden in Fettzellen – Wirkungen). Katecholamine– (Epinephrin, Isoprenalin).
2) Wassereinlagerungen in den Nieren – Auswirkungen Vasopressin;
3) Aufrechterhaltung der Ca +2-Homöostase – Wirkung der Nebenschilddrüsenhormone;
4) eine Erhöhung der Häufigkeit und Stärke der Kontraktionen des Herzmuskels – die Wirkung von Katecholaminen (Epinephrin, Isoprenalin)
5) Regulierung der Steroidbiosynthese in den Nebennieren und Gonaden – die Wirkung von Corticotropin oder follikelstimulierendem Hormon;
6) – Entspannung der glatten Muskulatur und vieler anderer hormoneller und nervöser Prozesse.
Wenn der neuronale oder hormonelle Reiz abgeschlossen ist, werden die intrazellulären Wirkungen von cAMP durch die Aktivierung des cAMP-abbauenden Enzyms beendet.
Einer der Mechanismen der therapeutischen Wirkung Koffein, Theophyllin und anderen Methylxanthinen ist die Hemmung des cAMP-Abbaus.
2) Ca +2 und Phosphoinositide.
Einige Hormone, Neurotransmitter und Wachstumsfaktoren binden an einen Rezeptor auf der Oberfläche der Effektorzelle. Das Signal wird an das G-Protein weitergeleitet. Anschließend wird Phospholipase C aktiviert. Diese spaltet gezielt Phospholipide der Plasmamembran unter Bildung von zwei sekundären Botenstoffen: 1) Diacylglycerin, 2) Inosittriphosphat.
Diacylglycerin aktiviert die Proteinkinase C, die Enzyme phosphoryliert und ihre Aktivität verändert.
Inosittriphosphat setzt Ca 2+ aus intrazellulären Speichern (sarkoplasmatisches Retikulum, Mitochondrien) frei. Ca 2+ verändert Zellfunktionen. Es provoziert beispielsweise Muskelkontraktionen usw.).
Lithium, das zur Behandlung manisch-depressiver Zustände eingesetzt wird, wirkt über Phosphoinositide.
3) cGMP. Im Gegensatz zu cAMP ist es nur in bestimmten Zelltypen an der Signalübertragung beteiligt. In der Darmschleimhaut und der glatten Gefäßmuskulatur funktioniert es parallel zum cAMP-System (als Reservesystem). Der Wirkungsmechanismus von cGMP wird auch durch Proteinphosphorylierung vermittelt.
Eine erhöhte cGMP-Konzentration bewirkt eine Entspannung der glatten Gefäßmuskulatur aufgrund der Dephosphorylierung der leichten Myosinketten.
Phosphorylierung: allgemeiner Mechanismus.
Fast alle Signaltransduktionsmechanismen über Second Messenger beinhalten Phosphorylierung.
Im Laufe der Evolution hat der Körper keine speziellen Rezeptoren für Medikamente entwickelt. Sie wirken über Rezeptoren für Neurotransmitter und Hormone. Fast alle Medikamente (möglicherweise mit Ausnahme der Vollnarkose) entfalten ihre Wirkung über Rezeptoren.
Wir haben die in der Plasmamembran der Zelle eingebauten Rezeptoren im Detail untersucht. Aber es gibt noch andere Arzneimittelrezeptoren. Grundsätzlich handelt es sich bei einem Rezeptor um etwas, an das ein Medikament im Körper bindet (interagiert). Albumin ist beispielsweise ein Rezeptor für Medikamente, die daran binden. Dieser Rezeptor ist jedoch nicht aktiv und führt nicht zu einer pharmakologischen Wirkung.
Zu den weiteren Klassen von Arzneimittelrezeptoren gehören:
1) Enzyme, 2) Transportproteine, 3) Strukturproteine.
Wenn sie an Medikamente gebunden sind, können sie gehemmt oder (seltener) aktiviert werden. Beispielsweise ist Dihydrofolatreduktase ein Rezeptor für Methotrexat.
Transportproteine(zum Beispiel Membranrezeptor für Herzglykoside – Na +, K +, ATPase).
Strukturproteine(zum Beispiel Tubulin – ein Rezeptor für entzündungshemmende Medikamente Colchicin).
Bei jeder Wechselwirkung eines Medikaments mit einem Rezeptor entsteht ein Medikament-Rezeptor-Komplex, der zu einer Veränderung des Stoffwechsels in Zelle und Organ führt. Es entsteht eine pharmakologische Wirkung. Sein Wert ist proportional zur Anzahl der Wirkstoff-Rezeptor-Komplexe.
Als Arzneimittel werden Arzneimittel bezeichnet, deren Wirkung mit der Stimulation von Rezeptoren verbunden ist Agonisten. Agonisten sind: 1) vollständig (verursachen eine maximale Reaktion) und 2) partiell. Letztere binden an Rezeptoren und erregen diese. Allerdings ist die pharmakologische Wirkung schwächer als die eines natürlichen Regulators. Substanzen, die die Wirkung bestimmter Agonisten beeinträchtigen, werden Antagonisten (Blocker) genannt.
Rezeptoren werden nach ihrer Empfindlichkeit gegenüber natürlichen Mediatoren und ihren Antagonisten klassifiziert. Beispielsweise werden Rezeptoren, die gegenüber Acetylcholin empfindlich sind, als cholinerge bezeichnet, und solche, die gegenüber Adrenalin (Adrenalin) empfindlich sind, als adrenerge.
Um die Rezeptoren anzuregen und die entsprechende Wirkung zu erzielen, werden sowohl die Mediatoren selbst (Noradrenalin, Dopamin und andere) als auch Medikamente verwendet, die eine Affinität zu den Rezeptoren haben. Letztere sind meist strukturelle Analoga von Mediatoren.
Manche Substanzen erregen den entsprechenden Rezeptor nicht durch direkte Interaktion mit ihm, sondern durch die Freisetzung von Mediatoren aus einer gebundenen (physiologisch inaktiven) Form oder durch die Hemmung von Enzymen, die Mediatoren zerstören.
Rezeptoren besetzen einen kleinen Teil der äußeren Zellmembran. Somit machen die Bereiche der Membran, die auf Acetylcholin reagieren, nur 1/6000 der gesamten Zelloberfläche aus
2. EINFLUSS AUF DIE ENZYMAKTIVITÄT. Die Wirkung einiger Medikamente beruht auf der Aktivierung oder Hemmung von Enzymen. PHYSOSTIGMINE hemmt beispielsweise die Aktivität der Cholinesterase, die Acetylcholin zerstört. Es verursacht Effekte, die für die Erregung des parasympathischen Nervensystems charakteristisch sind.
Einige Medikamente können eine Induktion bewirken, also den Gehalt des Enzymproteins erhöhen. Gleichzeitig nimmt ihre Aktivität zu. Beispielsweise reduziert Phenobarbital durch die Erhöhung der Aktivität der UDP-Glucuronyltransferase die Hyperbilirubinämie.
3. PHYSIKALISCHER UND CHEMISCHER EINFLUSS AUF ZELLMEMBRANEN.
Bei einigen Arzneimitteln ist die Natur der Zielmoleküle unbekannt. Ihre Wirkung ist nicht an bestimmte Rezeptoren gebunden. Vollnarkosemittel wirken beispielsweise, indem sie den Ionentransport verändern. Die therapeutische Wirkung von Salben, Pulvern und flüssigen Salben ist physikalischer Natur. Sie schützen die betroffenen Hautpartien oder Schleimhäute vor Reizungen.
Medikamente, die auf Rezeptoren wirken, lösen aus zwei Gründen ein breites Spektrum an Gewebe- und Systemreaktionen aus. Erstens haben verschiedene Gewebe unterschiedliche Rezeptoren. Zweitens haben verschiedene Rezeptortypen unterschiedliche Strukturen und dementsprechend unterschiedliche Funktionen, sodass die zelluläre Reaktion auf die Rezeptoraktivierung (Transduktion) je nach Struktur des Rezeptors erheblich variiert. Es gibt vier Arten von Rezeptoren, die sich in ihren kinetischen Eigenschaften unterscheiden:
G-Protein-gekoppelte Rezeptoren;
DNA-gebundene Rezeptoren;
Rezeptoren mit Tyrosinkinase-Aktivität (Tyrosinkinase-Rezeptoren);
Rezeptorgekoppelte Kanäle (RCCs).
Tyrosinkinase-Rezeptoren und RSCs Sie unterscheiden sich von den anderen dadurch, dass sie für die Manifestation einer zellulären Reaktion keine zelluläre Transduktion von Komponenten erfordern, wenn sie durch einen Agonisten aktiviert werden. Daher spielt das Rezeptormolekül eine größere Rolle als die molekulare Verbindung zwischen dem Arzneimittel und der Transduktion. Bei der Rezeptortyrosinkinase handelt es sich eigentlich um ein membrangebundenes Enzym, das durch einen Agonisten „angeschaltet“ wird. RCK ist ein spezifischer Ionenkanal, der sich strukturell von durch Medikamente oder Neurotransmitter gesteuerten spannungsgesteuerten Kanälen dadurch unterscheidet, dass er an eine hochspezifische Ligandenbindungsstelle und nicht an eine bestimmte spannungsempfindliche Stelle bindet. Bei RSC sind das Ligandenbindungszentrum und der Kanal funktionell unterschiedliche Regionen desselben Moleküls. RCK wird auch als ligandengesteuerter Ionenkanal bezeichnet. Die obige Klassifizierung ist praktisch, da sie Tyrosinkinasestrukturen und CSCs als Rezeptoren berücksichtigt.
G-Protein-gekoppelte Rezeptoren
G-Proteine sind Bestandteile der Transduktion. G-Protein-gekoppelte Rezeptoren sind auf der Zellmembran lokalisiert und bestehen aus 7 Transmembranhelices (I-VII).
In Abwesenheit von Agonist Der Rezeptor ist an ein G-Protein gekoppelt, das den Rezeptor in einer inaktiven Konformation hält. Es selbst ist ein Komplex aus drei Untereinheiten (a, beta und y). Wenn der Rezeptor im Ruhezustand ist, sind die drei G-Protein-Untereinheiten und Guanosindiphosphat (GDP) fest an die G-Protein-Alpha-Untereinheit gebunden. Die Bindung des Agonisten führt zu Konformationsänderungen im Rezeptor, was wiederum eine Konformationsänderung im G-Protein verursacht, was zur Dissoziation von GDP von der Alpha-Untereinheit führt. Dadurch wird eine Abfolge von Reaktionen initiiert und die G-Protein-gekoppelte Rezeptortransduktion gefördert, die im Folgenden beschrieben wird.
Aus Komponenten Transduktion Adenylylcyclase steht in direktem Zusammenhang mit G-Proteinen und kommt im Körper am häufigsten vor. Das zyklische Nukleotid cAMP wird aus Adenosintriphosphat (ATP) durch das Enzym Adenylylcyclase synthetisiert. cAMP hat verschiedene biologische Wirkungen.
Lager hat Auswirkungen auf den Energiestoffwechsel, die Zelldifferenzierung, die Ionenkanalfunktion und kontraktile Proteine
Lager phosphoryliert intrazelluläre Proteine (normalerweise Enzyme) durch die Wirkung von cAMP-abhängigen Proteinkinasen. Diese Proteinkinasen aktivieren cAMP und phosphorylieren Aminosäuren und Threonin, wobei ATP als Phosphorquelle genutzt wird. Das Ergebnis der Phosphorylierung ist:
Aktivierung hormonsensitiver Lipasen;
Inaktivierung der Glykogensynthase;
Aktivierung der Phosphorylasekinase und Umwandlung inaktiver Phosphorylase in aktive, was zu einer beschleunigten Lipolyse, einer verminderten Synthese und einem beschleunigten Abbau von Glykogen führt;
Aktivierung von L-Typ-Ca2+-Kanälen und des sarkoplasmatischen Retikulums in Herzzellen aufgrund von Phosphorylierung, was die Kalziumproduktion erhöht.










